
Image created using Canva
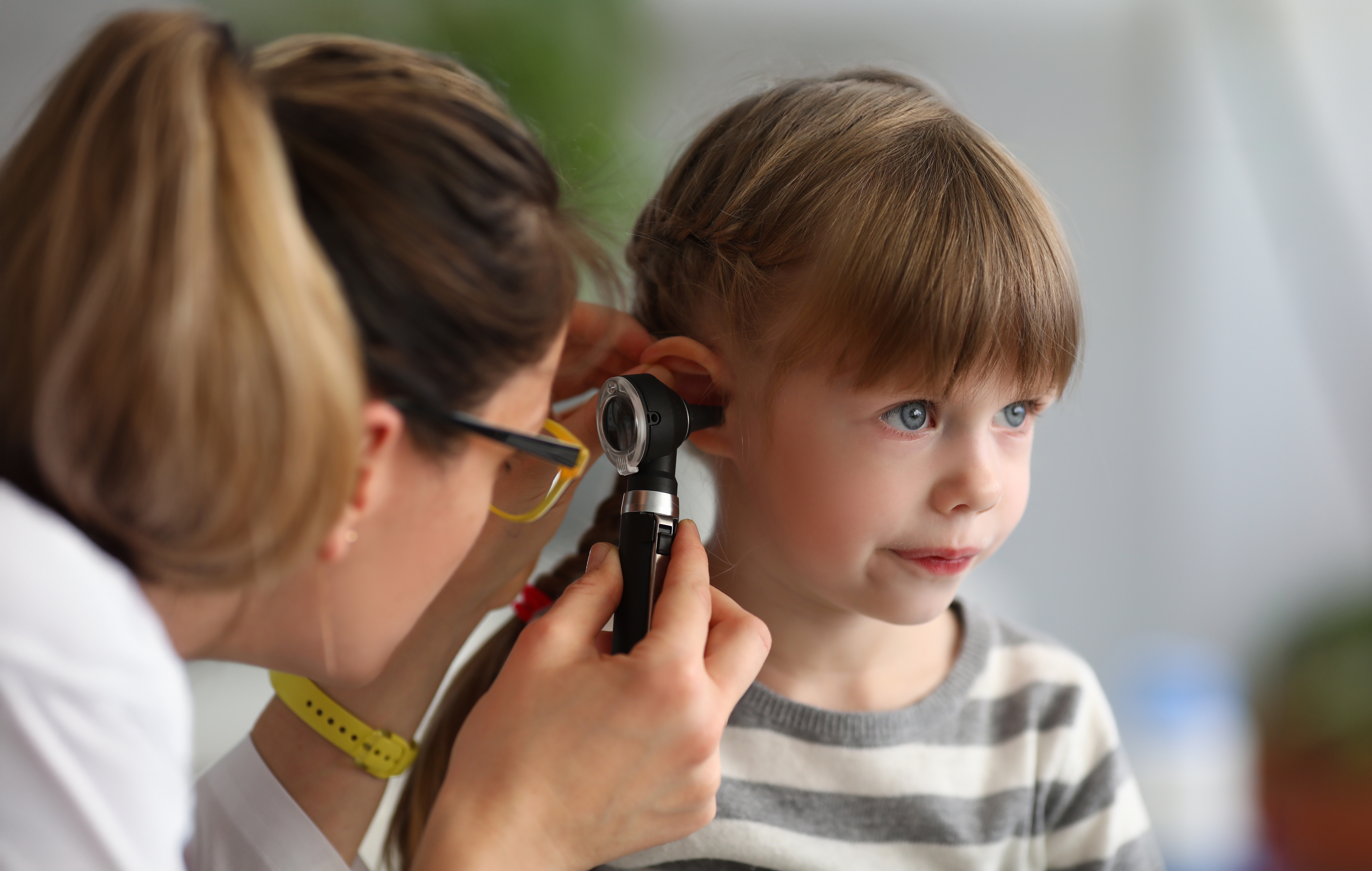
Mechanosensors in cars detect pressure, vibration, and movement. In our bodies, a similar role is played by tiny cells in the inner ear (cochlea) called hair cells. These specialized cells respond to the movement of fluid caused by sound, allowing us to hear.
Humans are born with around 15,000 hair cells in each cochlea. When these cells are damaged or die, they cannot be replaced, and this loss is a leading cause of permanent hearing loss.
Hair cells communicate with the brain by releasing a chemical signal that activates nearby nerve cells. One key molecule required for this process is otoferlin. Without it, hair cells cannot properly send sound information to the brain. Mutations in the otoferlin gene are a common cause of deafness present at birth.
Until recently, treatment options for people with otoferlin-related deafness have relied on cochlear implants. These devices bypass damaged hair cells by directly stimulating the auditory nerve. While cochlear implants can restore a sense of hearing, challenges remain, especially in understanding speech in noisy environments, hearing distant sounds, or appreciating music.
Now, gene therapy is changing what may be possible.
On April 23, 2026, the US Food and Drug Administration approved the first gene therapy to treat otoferlin-related deafness in children and adults. The therapy adds a healthy copy of the otoferlin gene to the hair cell that cannot make otoferlin. Otoferlin is delivered to hair cells using a virus, a small particle that efficiently enters hair cells without causing any adverse effects.
Several research teams are actively involved in clinical trials to test the safety and effectiveness of their otoferlin gene therapy. The overarching observation to date is that patients with otoferlin-related deafness who are treated with gene replacement recover usable hearing. It is heartwarming to see young children respond to their parents’ voices for the first time, dance to music, and learn the sounds that animals make. Remarkably, in one clinical trial, sensitive hearing was preserved in several patients 2.5 years after gene therapy, suggesting that the therapy is durable and that one dose may be all that is needed. The expectation is that additional gene therapies for other forms of deafness will emerge in the near future.
Otoferlin gene therapy is a stunning clinical breakthrough that would not have succeeded without nonhuman primate research. The safety of the surgical approach to the cochlea and the gene therapy virus delivery strategy must be assessed. The virus’s ability to access cells in the inner ear that are not the targeted hair cells must be defined. The ability of the virus to spread beyond the inner ear and infect the brain, liver, and other organs must be determined. The toxicity of the virus, or its ability to cause cell or tissue damage, must be described for the inner ear and dozens of tissues throughout the body. The safety, biodistribution, and toxicity studies that supported transition of the otoferlin gene therapy to clinical trials were each conducted in nonhuman primates. Nonhuman primate research will remain a centerpiece in translating basic science research into safe, successful human clinical trials that improve human health.
Related Research in Oregon
Oregon has played a steady role in the basic and translational science that makes these therapies possible. A few examples of related work across Oregon Health & Science University and the Oregon National Primate Research Center (ONPRC):
- Building rhesus macaque models of otoferlin-related deafness. A collaboration led by the Oregon Hearing Research Center and ONPRC is working to identify and study rhesus macaques deficient in otoferlin, the same protein at the center of the recently approved gene therapy. The goal is to evaluate gene therapy delivery, safety, and biodistribution in a model whose anatomy and biology closely resemble our own, bringing together hearing scientists, geneticists, and reproductive biologists from across OHSU. Read more: Gene therapy research to explore hearing restoration in monkeys (OHSU News, 2021)
- Restoring hearing before birth. A complementary line of work in the Oregon Hearing Research Center explores whether gene-correcting drugs delivered to the developing fetal inner ear could prevent congenital hearing and balance disorders such as Usher syndrome. Initial proof-of-concept studies in mouse models restored hearing and balance, and researchers expect that nonhuman primate studies will be a key next step before potential human application. *Read more: *New OHSU research suggests in-womb gene correction (OHSU News, 2020)
- Gene therapy for sensory disorders, a parallel path. As gene therapy begins to restore hearing, Oregon scientists at ONPRC and OHSU’s Casey Eye Institute have been advancing gene therapy for inherited forms of blindness, including recent work using lipid nanoparticles to deliver mRNA to the retina safely. Human clinical trials for macular degeneration are already underway. The parallels matter: the inner ear and the retina are both small, intricate sensory tissues, and lessons learned in one often inform approaches in the other. Read more: Nanotechnology may improve gene therapy for blindness (OHSU News, 2023)
- A view from inside the field. In a 2024 commentary in Molecular Therapy, this article’s author, John V. Brigande, Ph.D., reflected on the first published clinical results restoring hearing in congenitally deaf children, and on the long arc of basic, translational, and primate research that brought the field to that moment. Read more: Otoferlin gene therapy restores hearing in deaf children (Brigande, 2024, Molecular Therapy)